MaxSBM
1 Sequence based modifications - MaxSBM
To enable the detection of peptides containing sequence-based modifications (SBMs), like SUMOylation and ubiquitination, we configured the software to support SBMs via the “MaxSBM” module. SBMs are only supported for data-dependent acquisition (DDA) searches. The module enables identification of large complex PTMs that consists of monomers, and facilitates multiple fragmentation points and neutral losses.
The module is described in detail in Lennartsson et al., 20251.
1.1 Configuration
The configuration of MaxSBM is implemented in the graphical user interface (GUI). The modification configurations are found in the modification list, under the “configuration” tab (Figure 1.).
The MaxSBM module can be activated in MaxQuant and Andromeda by incorporating a variable modification in a search with the modification type “Sequence based modifier”. For SBMs, fragmentation masses are calculated automatically when providing the amino acid sequence of the SBM modification as input in the box under the “Composition” box (Figure 1.).
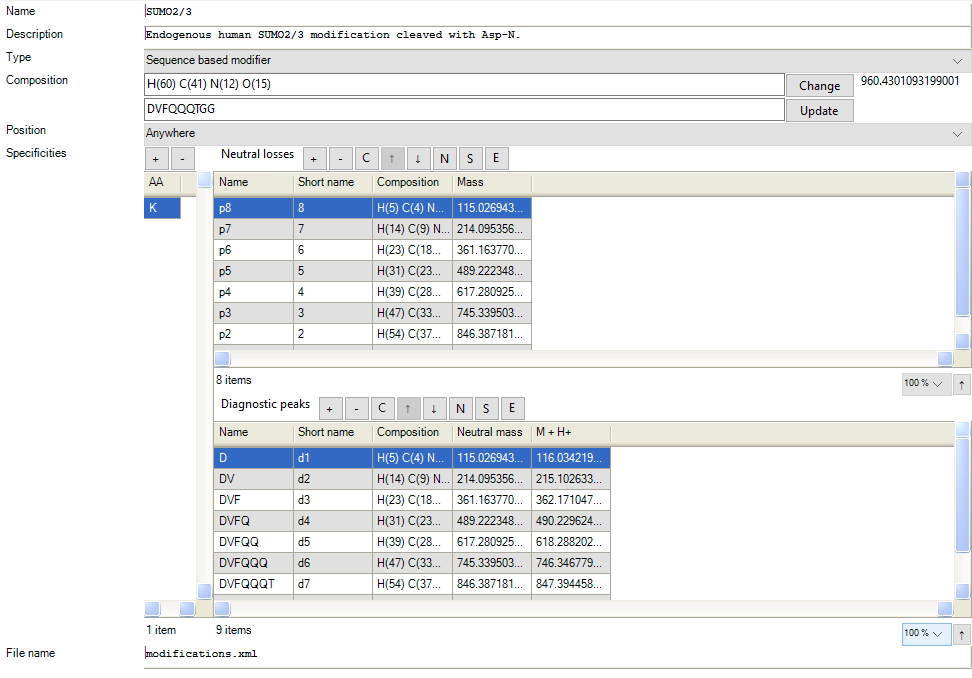 Figure 1. The configuration for including a SBM in a search is done by including a variable modification with the type “Sequence based modifier”, and defining a set of neutral losses and diagnostic ions. The diagnostic peaks and neutral losses can be calculated automatically by defining an amino acid sequence in the GUI.
Figure 1. The configuration for including a SBM in a search is done by including a variable modification with the type “Sequence based modifier”, and defining a set of neutral losses and diagnostic ions. The diagnostic peaks and neutral losses can be calculated automatically by defining an amino acid sequence in the GUI.
The PTM remainder masses are automatically calculated and defined in the list of neutral losses and diagnostic peaks, which the user can afterwards edit to optimize the search. SBM fragments that remain attached to the target peptide are denoted p-ions, based on their remaining PTM fragment. The p-ions remain attached to b- and y-ions from the peptide backbone itself, generating b-p and y-p ions. The remainder of the SBM is considered to be neutrally lost. Should the detached part of the SBM carry charge, these are annotated as diagnostic d-ions, which do not benefit sequencing of the target peptide backbone, but can be an indicator of the presence of the SBM in the fragment spectrum.
In the GUI, the p-ions are defined in the list of neutral losses. The mass of the p-ion is defined by the corresponding loss of the PTM. For example in Figure 1., p8 is highlighted in the list, which has the mass of the corresponding d-ion d1. This is because, p8 is calculated by subtracting d1 from the PTM full mass. In Andromeda, the p-ion is defined in the theoretical spectra by subtraction from the PTMs full mass in the search. The diagnostic ions masses, on the other hand, are defined as the mass of the d-ion.
The list of d-ions and p-ions can be modified by the user. The user can modify the naming of the ions and the numbering. The “name” that can be given is the long name of the fragment. The “shortname” is defining the naming and numbering of the fragments in the output and the viewer. For the p-ions, a number can be defined in the “shortname” which will be read and saved throughout the search. Here the user can add custom numbering. The field can also be left blank, where the ions will be numbered automatically.
1.2 Search space size
Since the number of added p-ions in the neutral loss list increases the search space substantially, we recommend the addition of these to be limited for increased identification rates. Several searches can be performed with different amounts and combinations of p-ions to iteratively determine the optimal configuration for your SBM.
1.3 Result
After the search is finished, the identified p-ions (as attached to either a b- or y-ion) are shown in the MaxQuant spectral viewer. The identified p-ions are reported with annotation and mass in the “msms.txt” search output table. The MaxSBM output with the complete set of modification identifications are found in the modification output table “sites.txt”. We recommend further processing with the downstream proteomics analysis platform Perseus.
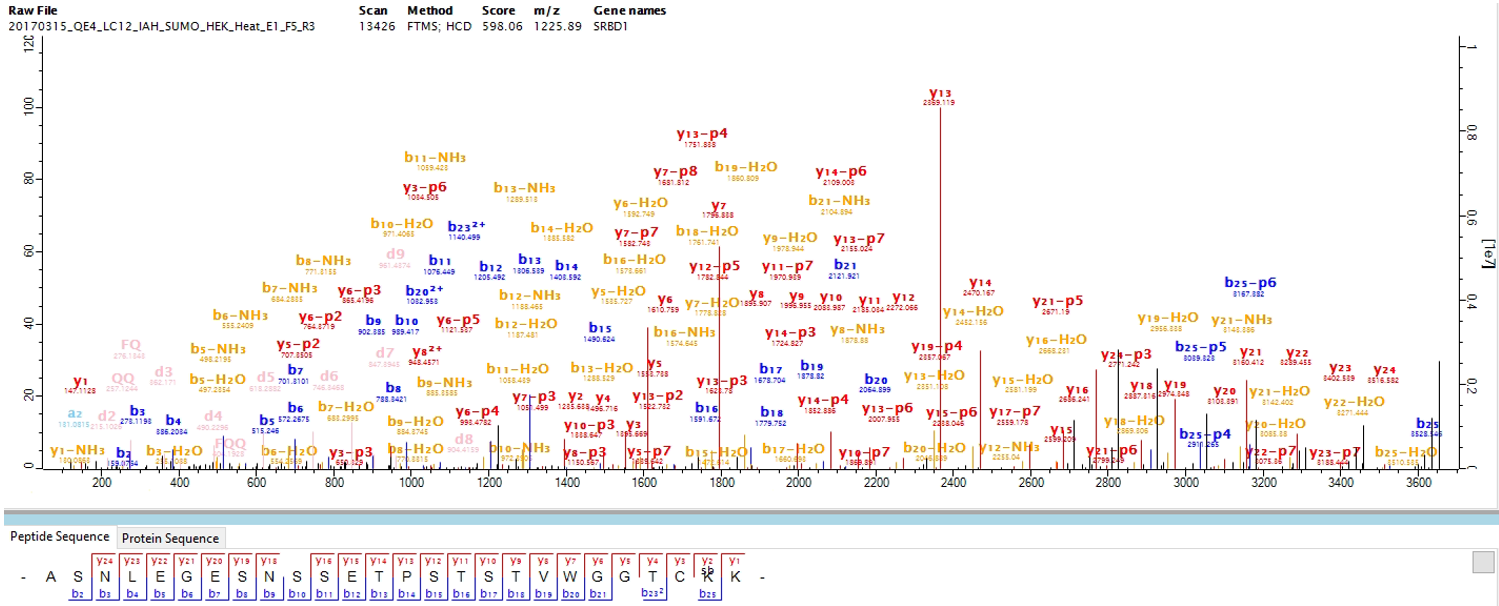 Figure 2. An example of an identified SUMOylated peptide in the MaxQuant viewer, using the MaxSBM workflow.
Figure 2. An example of an identified SUMOylated peptide in the MaxQuant viewer, using the MaxSBM workflow.